



A team of researchers has identified genomic variants that cause disabling pansclerotic morphea (DPM), a rare, severe inflammatory skin disorder, and report that the Janus kinase (JAK) inhibitor ruxolitinib may be a useful therapy, especially in patients who have not responded to other interventions.
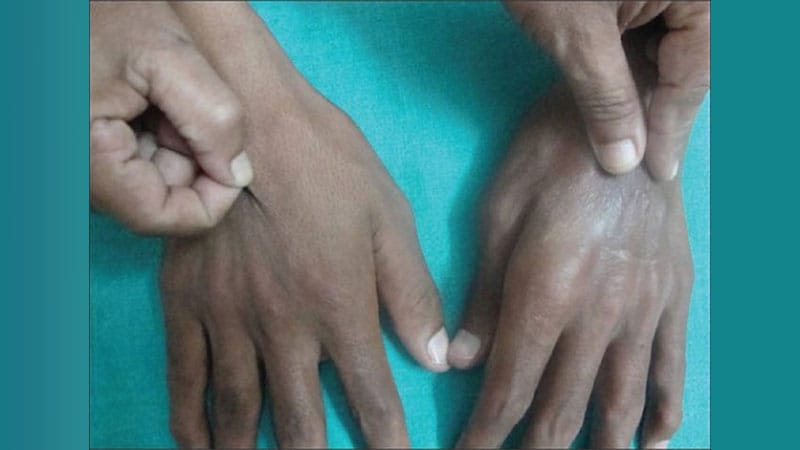
DPM was first reported in 1923, and while a genetic cause has been suspected, it had not been identified until now. The disease is the most severe form of deep morphea, which affects individuals with juvenile localized scleroderma. Patients, generally children under age 14, experience rapid sclerosis of all layers of the skin, fascia, muscle, and bone. DPM is also deadly: Most patients do not live more than 10 years after diagnosis, as they contract squamous cell carcinoma, restrictive pulmonary disease, sepsis, and gangrene.
In the study, published in the New England Journal of Medicine, the researchers discovered that people with DPM have an overactive version of the protein STAT4, which regulates inflammation and wound healing. The scientists studied four patients from three unrelated families with an autosomal dominant pattern of inheritance of DPM.
“Researchers previously thought that this disorder was caused by the immune system attacking the skin,” Sarah Blackstone, a predoctoral fellow in the inflammatory disease section at the National Human Genome Research Institute and co–first author of the study, said in a statement from the National Institutes of Health describing the results. “However, we found that this is an oversimplification, and that both skin and the immune system play an active role in disabling pansclerotic morphea,” added Ms. Blackstone, also a medical student at the University of South Dakota, Sioux Falls.
The overactive STAT4 protein creates a positive feedback loop of inflammation and impaired wound-healing. By targeting JAK, the researchers were able to stop the feedback and patients’ wounds dramatically improved. After 18 months of treatment with oral ruxolitinib, one patient had discontinued all other medications, and had complete resolution of a chest rash, substantial clearing on the arms and legs, and global clinical improvement.
The authors said that oral systemic JAK inhibitor therapy is preferred over topical therapy. Their research also suggested that anti–interleukin-6 monoclonal antibodies – such as tocilizumab, approved for indications that include rheumatoid arthritis and systemic sclerosis–associated interstitial lung disease, “may be an alternative therapy or may be useful in combination with JAK inhibitors in patients with DPM,” the authors wrote.
Most current DPM therapies – including methotrexate, mycophenolate mofetil, and ultraviolet A light therapy – have been ineffective, and some have severe side effects.
“The findings of this study open doors for JAK inhibitors to be a potential treatment for other inflammatory skin disorders or disorders related to tissue scarring, whether it is scarring of the lungs, liver or bone marrow,” Dan Kastner, MD, PhD, an NIH distinguished investigator, head of the NHGRI’s inflammatory disease section, and a senior author of the paper, said in the NIH statement.
“We hope to continue studying other molecules in this pathway and how they are altered in patients with disabling pansclerotic morphea and related conditions to find clues to understanding a broader array of more common diseases,” Lori Broderick, MD, PhD, a senior author of the paper and an associate professor at University of California, San Diego, said in the statement.
The study was led by researchers at NHGRI in collaboration with researchers from UCSD and the University of Pittsburgh. Researchers from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute of Allergy and Infectious Diseases also participated.
The study was supported by grants from the American Academy of Allergy, Asthma, and Immunology Foundation; the Ludwig Institute for Cancer Research; the University of California, San Diego, department of pediatrics; and the Novo Nordisk Foundation. Additional support and grants were given by the Deutsche Forschungsgemeinschaft, various institutes at the NIH, the California Institute for Regenerative Medicine, the Hydrocephalus Association, the Scleroderma Research Foundation, the Biowulf High-Performance Computing Cluster of the Center for Information Technology, the Undiagnosed Diseases Program of the Common Fund of the Office of the Director of the NIH, and the NIH Clinical Center.
This article originally appeared on MDedge.com, part of the Medscape Professional Network.
Source: Read Full Article