




The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for coronavirus disease 19 (COVID-19), first emerged in Wuhan, China in late December 2019, before spreading to the rest of the world.
Many governments were forced to enact costly and restrictive measures in an attempt to reduce the rapid transmission of SARS-CoV-2, from mandatory face masks and social distancing restrictions to full lockdowns and stay-at-home orders. While mass vaccination programs have allowed many developed nations to withdraw these restrictions, the COVID-19 pandemic continues to affect millions of people.
In a recent study published on the preprint server medRxiv*, researchers investigate the association between seasonal flu changes and gene expression to determine the ‘within-host’ immunity concept, with hopes that their findings will have ramifications for understanding COVID-19.

Study: Trajectories of gene expression, seasonal influenza, and within-host seasonal immunity: transfer value to covid-19. Image Credit: Gorodenkoff / Shutterstock.com
About the study
Microarray expression data was gathered from the Norwegian Women and Cancer Study (NOWAC). The study population consisted of women randomly selected from the Norwegian population.
A total of 172,000 invites and questionnaires were sent out, and those who accepted and participated were followed up through linkage to national cancer and death registries. Additional questionnaires were sent out every four to six years. A subset of these individuals was then invited to help establish a biobank for analyses of functional genomics, which included a questionnaire and blood sample collection.
Taken together, about 50,000 women participated in NOWAC. In the current study, 425 women who had not been diagnosed with cancer were used as controls in the discovery population, and 432 women who had been controls in a study examining the changes in gene expression following a breast cancer diagnosis were included in the replication population.
Study findings
Model A was created with a singular seasonal term and applied to the discovery dataset. To this end, 2,942 out of 6,118 possible genes were significant, 416 of which showed a log fold change above 0.2. Clear seasonal effects were observed in both winter and summer.
Up to 5% of all sick leaves were due to influenza-like illnesses (ILI), with the maximum value varying between the years but often reaching a peak during the colder months.
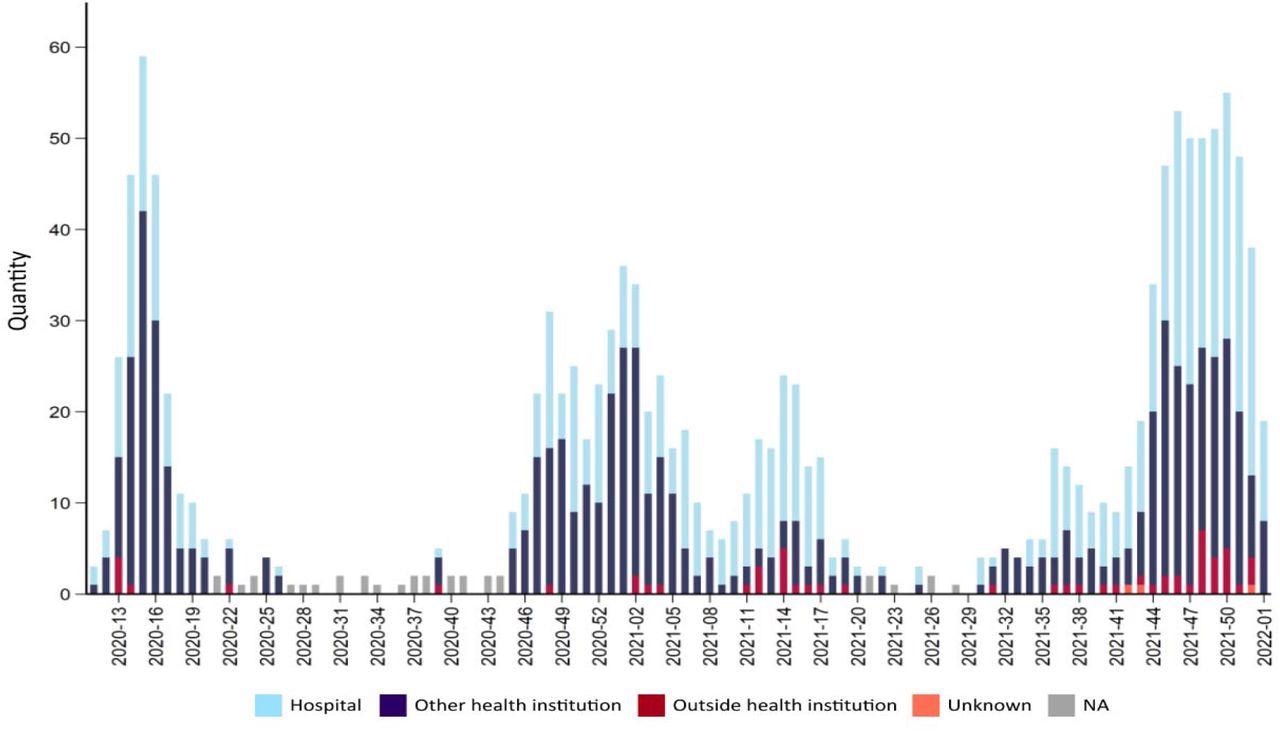
Number of deaths due to or with covid-19 for each week during the pandemic 2020-2022. Source FHI Weekly report 2022
Model B showed that most of the same genes, 1,983 in total, showed a significant seasonal or flu term when the model only included one of these variables. Moreover, 959 genes only have a significant seasonal term, while 611 only have a flu term. These terms mostly describe the same variability, with the seasonal term showing the flexibility to set maximum gene expression at any data in the year.
A total of 1,051 genes were associated with both a seasonal and flu term, with genes reduced in the flu season more likely to show a positive seasonal term, and the inverse remaining true for genes that rose during flu seasons. Six genes were selected with the most significant seasonal and flu effects, three of which showed reduced expression during flu season, whereas the remaining three were associated with an increased expression. The three genes with increased expression showed a maximum seasonal trend during the summer, with the reduced expression showing a minimum trend in the summer.
Model C had both a seasonal and flu term, which was used for moving flu intensities trend over days in order to determine whether the changes in gene expression were before, during, or after the changes in flu intensity. To this end, the number of genes with significant seasonal terms and no flu term was lowest when the flu term was five days later than the actual data, thereby indicating that the main change occurs before the rise in flu sick leaves. This is further supported by results showing that the most downregulated genes reached their minimum negative weight at around -20 days.
Replication analysis was performed to reduce noise in the data before interpretation. A total of 87 genes from the discovery dataset could not be found; however, the remaining 658 genes had either a significant season or flu term, whereas 369 had both. Again, clear seasonality was discovered.
Following the removal of genes with an unknown function, correlation analysis identified another 22 genes with more than one season's difference in the estimated month of maximum, which was also excluded. This resulted in Pearson’s correlation coefficient increasing from 0.83 to 0.92.
Conclusions
The authors of the current study highlight that they have discovered a novel covariation between seasonal influenza and annual changes in gene expression for a specific set of genes in immune cells. While this may be expected, with gene expression naturally changing in immune cells during infection, these findings support further investigations into within-host seasonal immunity, as this concept is currently underdeveloped and underinvestigated.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information
- Lund, E., Holden, M., Busund, L. R., et al. (2022). Trajectories of gene expression, seasonal influenza, and within-host seasonal immunity: transfer value to covid-19. medRxiv. doi:10.1101/2022.03.01.22271679. https://www.medrxiv.org/content/10.1101/2022.03.01.22271679v1.
Posted in: Genomics | Medical Science News | Medical Research News | Disease/Infection News
Tags: Blood, Breast Cancer, Cancer, Cancer Diagnosis, Coronavirus, covid-19, Flu, Gene, Gene Expression, Genes, Genomics, immunity, Influenza, Microarray, Pandemic, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Virus

Written by
Sam Hancock
Sam completed his MSci in Genetics at the University of Nottingham in 2019, fuelled initially by an interest in genetic ageing. As part of his degree, he also investigated the role of rnh genes in originless replication in archaea.
Source: Read Full Article